
Azərbaycanda da rast gəlinən xromosom anomaliyası - Di George Sindromu nədir?
- 2021.02.16 15:37
- 3861 Baxış

DG sindromu 22 ci xromozomun 22q11.2 delesiyası və ya translokasiyası nəticəsində xromozomun yenidən şəkillənməsi ilə meydana gələn genetik bir anomaliyadır. Bu bölgədəki TBX1 geni xəstəlik yaratmaqla önəmli rol oynayır.
Bu sindromun prevalansı (yəni sindromun toplumda özünü biruzə vermə sıxlığı) təxminən 1:4000 ilə 1:6000 arasıdır. Pasiyentlərin böyük bir qismində 22q11.2 mikrodelesiyası görülməklə yanaşı bəzən sporadik de nova mutasiya ilə də meydana çıxa bilir.
Təxminən 6-7 % hallarda ailəvi irsiyyət tipi görülür. Di George sindromu autozom dominant ötürülmə ilə seyr edilir.
Bu sindrom əsas olaraq hamiləliyin 3-8.ci həftələri arasında meydana çıxan qüsurlu inkişaf nəticəsində yaranır. 22 ci xromozomdakı gen defekti nəticəsində bətndaxili inkişafın 3.cü və 4.cü faringal yarıqların anormal inkişafı olur. Bu sindrom timus hipoplaziyası, ikincili immun çatışmamazlıq, anadan gəlmə ürək və böyük damar anomaliyası, spesifik üz quruluşu, inkişaf geriliyi, hipoparatiroidizm
kimi klinik əlamətlər ilə özünü biruzə verir.
22q11 mikrodelesiya sindromu olan pasiyentlərin 75 % də ürək anomaliyası aşkarlanır və bu pasiyentlərdə ölümə səbəb olur.
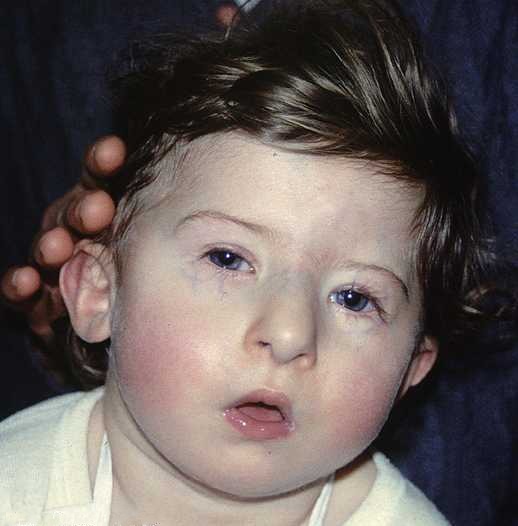
Üz anomaliyası; kiçik çənə, qulaqların nisbətən aşağı pozisiyada olması, gözlər arası məsafənin çox olması vb. kimi əlamətlərlə özünü biruzə verir.
Di George sindromunda timus vəzinin zəif inkişaf etməsi vəya yaranmaması səbəbindən T hüceyrələri sintez olunmur və immun sistem çatışmamazlığına səbəb olur. Timus funksiyası olmayan və sümük kök hüceyrəsində T hüceyrələrə çevrilə bilmədiyi uşaqlar 2 yaşına qədər ağır infeksiyon səbəbindən ölürlər. Bu zaman ediləcək ən doğru addım T hüceyrələrinin köçürülməsidir. Prosedur anesteziya
altında aparılır və klinik ədəbiyyatda yaşam şansının 75% olduğu bildirilmişdir.
Paratiroid vəzi parathormon ifraz edərək Ca balansını tənzimləyir. Di George sindromlu xəstələrdə bu vəz olmadığından buna bağlı olaraq Ca çatışmamazlığı yaranır və qıcolmalar görülə bilər.
Əlavə olaraq, onu da qeyd etmək lazımdır ki, bəzi xəstəliklər vardır ki, DG sinromu ilə bənzər simptomları göstərir. Məsələn,
Smith-Lemli-Opitz sindromu (polidaktiliya və damaq yarığı)
Goldenhar sindromu (qulaq anomaliyaları, ürək anomaliyaları, böyrək
fəsadları)
Alagille sindromu
CHARGE sindromu
Klinik şübhə olduqda diaqnozu dəqiqləşdirmək üçün FISH, MLPA, SNP array, microarray, qPCR kimi metodlar tətbiq edilə bilir. Hamiləlik zamanı klinik əlamətlər USM vasitəsi ilə də görülə bilər.
DG sindromlu pasiyentlər üçün spesifik bir müalicə sxemi yoxdur. Mqalicə sxemi əsasən klinik əlamətlərin səbəb olduğu simptomları aradan qaldırmağa əsaslanır. Ağır immun çatışmamazlığı olan pasiyentlərin xüsusilə infeksiyalardan qorunması, profilaktik tədbirlərin görülməsi, bu pasiyentlərə canlı virus peyvəndinin vurulmaması, Ca-vit D istifadəsi kimi tədbirlər görülür. İmmunaziyasiya venadaxili immunoqlobulin və antibiotik profilaktika rejimləri pasiyentin fərdi labarator dəyərlərinə əsasən 6-12 ay arası təyin edilir. Yarıq damaq halı kimi qüsurların cərrahiyyə metodu ilə düzəldilməsi uzman otoloringoloq, plastik cərrah və üz-çənə cərrahı tərəfindən qiymətləndirilir. Yarıq damaq probleminin düzəlməsi qidalanma qabiliyyətinin və danışıq qabiliyyətinin inkişaf etməsinə, pulmonar infeksiya hallarının isə azalmasına kömək edə bilər. Eşitmə problemi yaşayan pasiyentlər üçün isə audiometrik diaqnostika mütləqdir. Əlavətən bunu da qeyd etmək lazımdır ki, timus və hemopoetik transplantasiya olmadan bu pasiyentlər 12 aya qədər dünyasını dəyişirlər.
DG sindromlu övladı olan hər bir valideyinin bilməsi vacibdir:
Düzgün və dəqiq genetik konsultasiya
İnfeksiyanın ilkin əlamətləri
Hipokalsimiyanın əlamətləri
Cərrahi müdaxilə variantları
Qidalanma vəya dil çətinliyini aradan qaldırmaq üçün danışma terapiyası
Psixoloji yardım
Diaqnoz və idarə etmə çətin olsa belə düzgün mütəxəssislərin birgə səyi nəticəsində DG sindromlu pasiyentlərin ölüm hallarını azaltmaq olar.
⏺Analizə və genetik konsultasiyaya görə Afgen Genetik Diaqnoz Mərkəzinə müraciət edə bilərsiniz.
3 mkr., Pişəvəri 110 (Cavadxan küç 24), M. Əcəmi m.st. yaxınlığında;
☎️(+994 12) 430 35 17
????(+994 50) 265 66 00








































Ən Çox Oxunanlar




















Yeni Xəbərlər



















